Our Research
Mechanisms and Interventions of Folate-Nonresponsive Neural Tube Defects (NTDs) -Lead Scientist Dr. Richard Finnell
Although it is established that periconceptional use of folic acid prevents a significant percentage of the population burden of NTDs, the mechanisms underlying the processes by which this B vitamin reduces NTD risk remains unknown. Importantly, there are significant numbers of NTDs that are not preventable by folic acid supplementation, with these folate-resistant NTDs occurring at an apparent baseline rate of 5 per 10,000 live births (Heseker et al 2009). Thus, NTDs remain a substantial public health problem, and there is a critical need to understand the mechanisms underlying folic acid-resistant NTDs and to develop novel intervention strategies targeting this population. Our laboratory utilizes several mouse models of folate-resistant NTDs, such as mouse knockouts of Slc25a32, Mthfd1l, and Fkbp8 to study mechanisms contributing to NTDs. We also screen human NTD cases for genetic variants that may contribute to NTD risk, even in folate replete populations, and perform functional assays using mouse, cell culture, and biochemical models to determine how these variants contribute to NTD pathology in molecular, cellular, and tissue level systems.
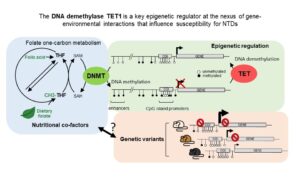
Epigenetic Regulation of Neural Tube Closure Defects -Lead Scientist Dr. Kian P. Koh
Most human traits and diseases are influenced by the complex interactions between one or more genes with environmental factors, such as nutrition or chemical pollutants. Thus, the impact of an environmental exposure on disease risk is different for people with different genetic make-up, or vice versa, the impact of a genotype on disease risk is different under different environmental conditions. To gain insights into these gene-environment interactions, we leverage on the role of the epigenome, which comprises chemical modifications of DNA and histones in chromatin that regulate gene expression, as an interface between the genome and the environment. The overarching goal of our research program is to identify mechanisms by which epigenetic regulators interact with nutrition-metabolites and genetic risk factors to influence neurodevelopment including neural tube closure. Our recent studies identify a critical role of the DNA demethylase TET1 in the interplay between folate-dependent one-carbon metabolism and genetic modifiers for neural tube defects (NTDs). By decoding the interplay of TET1 with the genome, metabolome, and epigenome, we shed light on genomic loci and mechanisms associated with FA responsiveness and NTD risk, implicating cellular trafficking and signaling pathways. Targeted intervention strategies tailored to rectify these pathways with individualized micronutrient supplementation plans may have a major impact in preventing all NTDs.
Imaging Studies of Dolutegravir Teratogenicity in an Organoid Model System -Lead Scientist Dr. Robert Cabrera
Recent data from a surveillance study in Botswana reported an increased risk of NTDs among newborns whose mothers were taking the HIV integrase inhibitor, dolutegravir, during the first trimester of pregnancy. Following these findings, the World Health Organization and the U.S. Food and Drug Administration issued recommendations and guidelines emphasizing the importance of considering the potential risks regarding the use of dolutegravir during pregnancy. In order to better understand the mechanisms by which this anti-retroviral medication may adversely impact embryonic development, we generated brain organoids, which are three-dimensional, miniaturized models of the human brain that can be used to study early neural development, and to assess the potential neurotoxicity of drugs and environmental compounds. We utilized human stem-cell-based brain organoids that were exposed to the drug and its impact on gene expression were determined via RNA sequencing in relation to structural information observed by Optical Coherence Elastography (OCE), Optical Coherence Tomography (OCT), and Brillouin microscopy. The RNA sequencing data indicated that dolutegravir modulates the expression of the gene networks required for brain organoid development. The Brillouin frequency shift observed in the surface of brain organoids indicates increased stiffness levels after exposing brain organoids to dolutegravir, while OCE measurements indicate decreased organoid volumes and a global stiffness decrease. With regard to mechanistic interactions of dolutegravir in brain organoids during neurodevelopment, since dolutegravir was previously reported to be a partial antagonist of Folate Receptor 1 (FOLR1), we focused on this nutrient transporter and observed increased FOLR1 expression in brain organoids, with increased stiffness levels specifically at the organoid surface and impaired growth and development.

Understanding Genetic Risk Factors that Contribute to Neural Tube Defects -Lead Scientist Dr. Richard Finnell
Although more than 300 genes have been associated with NTDs in mice, the genetic etiology of human NTDs remains poorly understood. We hypothesize that relatively few NTDs are caused by single gene variant(s), while the majority of NTDs are the result of digenic, or polygenic rare deleterious variants which work together through specific molecular and cellular processes. Our studies involve testing our hypotheses by analyzing large-scale (~1000) whole exome sequencing datasets of NTD patients and performing both in vivo and in vitro functional analysis to determine the underlying mechanisms by which identified gene variants interfere with neural tube closure. We hope that the genomic data will benefit our efforts for prevention and the treatment of spina bifida.
To realize these loft goals, we will initially identify NTD risk genes by analyzing whole exome DNA sequences of 500 NTD case-parent trios and 500 NTD sporadic cases. This will be followed by in vitro and in vivo functional analyses of variants for 2-3 novel NTD associated genes. We will also test whether digenic and trigenic variants identified in NTD patients are pathogenic in mice by creating transgenic animals and determining how the interaction of these variants results in failed neural tube dlosure.
Understanding the Genetic Complexity in Spina Bifida (HD111089) -Lead Scientist Dr. Richard Finnell
Among neural tube defects (NTDs), myelomeningocele (spina bifida:SB), is a devastating but survivable, human structural malformation. Up to 70% of SB cases are attributed to genetic predisposition, with intrauterine environment precipitating SB manifestation in those at risk. Despite decades of research into genetic factors that underlie NTDs in mouse models, translation to human risk assessment and amelioration of SB cases remain elusive. This is largely attributable to the limitations of the candidate gene approaches to genetic studies of human NTDs. Here, our comprehensive systems biology approach to mutation burden in whole genome sequence analyses is illuminating molecular pathways to human SB through genome interrogation of protein coding and non-coding regions, and introduces machine learning to select, in an untargeted fashion, genes with SB discriminatory potential based on gene enrichment by rare, likely deleterious protein coding variants.
In this collaborative research program with Prof. Betsy Ross at Weill Cornell Medical College, our laboratory will test the functional significance and interactions of newly detected candidate genes and variations by making double- and higher multiple-heterozygous variant interactions using existing and CRISPR-edited mice to examine the impact on neural tube closure. Our computational approaches conducted in New York are highly innovative in the field. Importantly, we use advanced technology for the functional testing of genetic variation and interactions associated with SB, including gene editing of human stem cells and isogenic controls, and mice for in vitro and in vivo modeling of consequences for key steps in early neural development, and evaluation of the cell-type gene expression changes induced by these variants. Insights from these studies will pave the way for a precision medicine capability to individualize NTD prevention and care for the hundreds of thousands of patients living with SB.
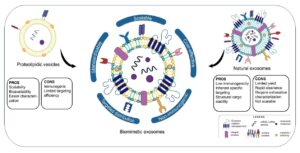
Biomimetic delivery systems for the prevention and treatment of congenital malformation (HD113702) -Lead Scientist Dr. Bruna Corradetti
Despite their well-established characteristics and potential, the application of exosomes in clinical settings is limited by intrinsic drawbacks in their specificity, production, and scalability. Alternatives to exosome include lipid nanoparticles, which have recently proven effective in clinical settings, and liposomes. In 2020, my research team developed a scalable and efficient cell extrusion-based approach to produce the so-called Immune-Derived Exosome Mimetics (IDEM) that showed a 2.5-fold increase compared with natural exosomes released by the same number of cells. IDEMs retain the distinctive physiochemical and molecular features of natural exosomes (i.e., CD63 and CD81, key characterizing markers), with an increased mechanical stability that correlates with an improved cellular uptake efficiency. As an effective biomimetic drug delivery system, IDEMs can be loaded with therapeutics, including drugs and RNAs. IDEMs loaded with doxorubicin showed an increase in drug encapsulation efficiency (28% vs 17%) and release overtime (up to 20% more) compared to natural exosomes, which were associated to incremental drug uptake by target cancer cells, in 2D and 3D culture systems, and to a more effective cytotoxic and apoptotic effect of DOXO-loaded particles compared to free drug. More recently, we have formulated biomimetic exosomes from amniotic fluid-derived mesenchymal stem cells, which are currently under investigation for their potential application as minimally invasive strategies for the prevention and treatment of congenital malformations.
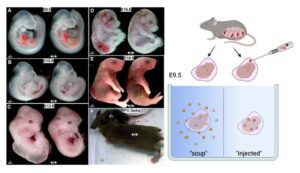
Make Stacy Walk-Lead Scientists Dr. Bruna Corradetti and Dr. Richard Finnell (HD113702)
One of the most ambitious programs ongoing in the laboratory involves utilizing cell free protein factors to help mitigate the co-morbidities associated with spina bifida in our FKBP8 knockout mouse model. This program draws upon the diverse research strengths of our birth defects research team beyond the lead scientists, including: Dr. Jacqueline Parchem, a maternal-fetal medicine specialist who provides amniotic fluid, placental and umbilical cord tissues from which stem cells and exosomes are isolated for the experiments. Dr. Bogdan Wlodarczyk and Dr. Ying (Linda) Lin perform the mouse surgeries to deliver the test compounds to the dam/embryos. They are currently exploring a number of different delivery methods, including i.p. injection, trans-uterus injection, embryo culture, transvaginal approaches, and direct fallopian tube injection. Drs. John Steele and Robert Cabrera devise the testing strategies, examine the embryos, and perform related neurobehavioral assays. These experiments draw upon decades of preclinical and clinical research evaluating mesenchymal stromal/stem cell (MSC) utility which have demonstrated their capacity to promote endogenous tissue repair and regeneration in acute and chronic injury and disease. As spina bifida can be diagnosed before irreversible neurological damage has occurred, utilizing stem cells or cell free therapies have been proposed as a novel intervention for treatment/repair of spina bifida. The hope is for both for the application of regenerating neural tissue and protecting tissue from further damage. Moderately successful application of several repair strategies has been employed in rat models of spina bifida, using both direct microinjection into the spina bifida lesion or injection into the amnion, and using both rat and human MSCs. Additionally, human MSCs have been used to repair spina bifida in sheep models resulting in improved locomotor outcomes and motor function. As our studies with the FKBP8 knockout mouse progress, we have access to a herd of sheep with a high spontaneous prevalence of spina bifida available for transitioning to a large animal spina bifida model.
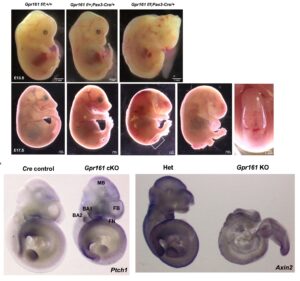
The role of Sonic Hedgehog Signaling Genes in the Etiology of Spina bifida -Lead Scientist Dr. Sung Eun Kim
The Sonic hedgehog (Shh) signaling is one of morphogen signaling involving neural tube morphogenesis. Several human spina bifida gene variants in Shh signaling have been identified; however, their genetic and developmental roles in spinal neural tube development have not been well understood. Our laboratory identified the spina bifida genetic variants of GPR161 and FKBP8, genes in Shh signaling and has established the mouse spina bifida models with both genes, which are folate resistant. Based on our observations, we are specifically interested in three big research questions. 1) The complex molecular genetics that these genes contribute to the onset of spina bifida in human and mouse models, 2) which cellular lineages are involved in GPR161 and FKBP8 mediated spinal neural tube development, and 3) the inter-relationship between Shh signaling genes and other morphogen signaling genes (Wnt and planar cell polarity) and genes in metabolic pathways in the etiology of spina bifida. We will utilize our lab-established mouse models and in vitro human-based neural tube systems and employ multi-omics experiments. This project will illuminate the novel genetic mechanisms that can be utilized in the development of mechanism-based intervention and therapeutic strategies to prevent folate resistant spina bifida.
Using Embryonic Stem Cells to Screen for Reproductive Toxicants -Lead Scientist Dr. Robert Cabrera
There are over 85,000 industrial chemicals presently registered with the federal government, the vast majority are inadequately studied with regards to their potential environmental and biological impacts. Over 30,000 of these industrial chemicals are sold at quantities exceeding 400 million tons per year; however, there also remains a considerable knowledge gap regarding the relationship between exposure to these industrial chemicals and possible adverse health effects. Many of these chemicals are released into the environment and can be absorbed via ingestion, inhalation, or dermal exposures. Additionally, these chemical can also be transported to the fetus after maternal exposure and can have adverse effects on fetal and neonatal health. This lack of information on industrial chemicals released into the environment leaves the federal government with an ever increasing number of chemicals that may contribute to immediate and long-term health effects in exposed animal and human populations, most of which need to be characterized and prioritized by their individual health risks. The Finnell Laboratory is working to develop a solution to this problem via high-throughput, high-information content biological screens on industrial chemicals. We hypothesize that high throughput screening (HTS) ESCs can recapitulate observations from in vivo models exposed to environmental pollutants. Further, in the absence of a priori toxicological data, we hypothesize that HTS of ESC can be used for prediction of in vivo impact, risk assessment, and prioritizing of chemicals for in vivo testing and epidemiological studies.
Investigating the Impact of Folic Acid Supplements on Cerebrospinal Fluid Folate Levels in Transgenic Mice -Lead Scientists Dr. Linda Lin and Dr. Bogdan Wlodarczyk
Folic acid, an essential nutrient absorbed by the intestine, plays a vital role in various physiological processes. Active transport, facilitated by the choroid plexus (CP) and key transporters such as Pcft, Folr1, and Rfc, is required for folates to reach the brain. Despite this, the ineffectiveness of folic acid (FA) supplementation in correcting cerebrospinal fluid (CSF) folate levels remains puzzling. This study aims to uncover the reasons behind this phenomenon and explore its potential connection with neurobehavioral changes in transgenic mice. The primary objective is to elucidate the mechanisms that hinder the normalization of CSF folate levels despite FA supplementation. Specifically, we will investigate the roles of Pcft, Folr1, Rfc, and other relevant genes. Additionally, we aim to establish any potential associations between altered CSF folate levels and neurobehavioral changes in transgenic mice.
Phenotypic Rescue of VPA-induced NTDs using inositol during key developmental periods -Lead Scientists Dr. Linda Lin and Dr. Bogdan Wlodarczyk
Valproic acid (VPA) usage during pregnancy is associated with an increased risk of neural tube defects. Inositol, a naturally occurring carbohydrate, has been proposed as an alternative supplement for use during pregnancy to reduce the prevalence of spina bifida. We are studying the potential of 3,4-dibutyryl-D-chiro-inositol to counteract the negative effects of VPA on fetal development. The primary objective is to assess the efficacy of inositol supplementation, specifically 3,4-dibutyryl-D-chiro-inositol, in reducing the risk of VPA-induced NTDs. The study aims to understand the underlying mechanisms, focusing on cellular signaling, neural development, and its influence on insulin signaling and glucose metabolism.